Medical Device Regulations in the People’s Republic of China -
the New Trend and the Challenges and Opportunities
to European Medical Device Manufacturers
Wissenschaftliche Prüfungsarbeit
zur Erlangung des Titels
„Master of Drug Regulatory Affairs“
der Mathematisch-Naturwissenschaftlichen Fakultät
der Rheinischen Friedrich-Wilhelms-Universität Bonn
vorgelegt von
Xinyu Meng
aus Jiangsu, China
Bonn 2019
Betreuer und 1. Referent: Dr. Ehrhard Anhalt
Zweiter Referent: Dr. Santiago Figueroa-Perez

TABLE OF CONTENTS
I
TABLE OF CONTENTS
TABLE OF CONTENTS .................................................................................. I
List of Abbreviations ................................................................................... III
List of Tables............................................................................................... IV
List of Figures ............................................................................................. IV
1. Background ......................................................................................... 5
1.1 Aims and objectives .............................................................................. 6
1.2 Notes .................................................................................................... 6
2. Marketing Approval System for Medical Devices in China ............. 7
2.1 Qualification of the applicant ................................................................. 7
2.2 “Agent” of Imported Medical Devices .................................................... 7
2.3 Classification of Medical Devices ......................................................... 9
2.4 Registration Units ............................................................................... 11
2.5 Marketing Approval Certificates .......................................................... 12
2.6 Label and IFU ..................................................................................... 13
3. Application Procedures for Marketing Approvals of Imported
Medical Devices in China ................................................................. 15
3.1 Application Types ............................................................................... 15
3.2 Application procedures for approval of class II and class III imported
medical devices .................................................................................. 15
3.2.1 Approval procedures (flowchart) ......................................................... 16
3.2.2 Application timeline ............................................................................. 17
3.2.3 Application requirements .................................................................... 18
3.3 Filing of class I imported medical devices........................................... 21
4. Discussion ........................................................................................ 23
4.1 Possible challenges from the current medical device regulations in
China .................................................................................................. 23
4.1.1 The importance of “Agent” .................................................................. 23
4.1.2 Possible classification differences ...................................................... 24
4.1.3 Possible difference in registration unit ................................................ 24
4.1.4 Possible issues with the Chinese label and IFU ................................. 25
4.1.5 Triggers for an “initial registration” ...................................................... 25
4.1.6 Possible issues with the submission dossier ...................................... 25

TABLE OF CONTENTS
II
4.2 Recent changes indicating the new trend in Chinese medical device
regulations .......................................................................................... 32
4.2.1 Clinical trial exemption ........................................................................ 32
4.2.2 Proposed clinical evaluation exemption for class II medical devices .. 35
4.3 Alternative options to registration testing ............................................ 36
4.4 Post market surveillance being strengthened ..................................... 39
4.5 Overseas on-site audits to be conducted ............................................ 40
4.6 Special review procedure for innovative devices ................................ 42
5. Conclusion and outlook ................................................................... 45
6. Summary ........................................................................................... 48
Acknowledgement ...................................................................................... VI
References ................................................................................................. VII

List of Abbreviations
III
List of Abbreviations
AIMDD
Council Directive 90 / 385 / EEC of 20 June 1990 on the
approximation of the laws of the Member States relating to
active implantable medical devices
CAPA
Corrective and Preventive Action
CFDA
China Food and Drug Administration
CMDE
Centre for Medical Device Evaluation (China)
EC
Ethics Committee
EU
European Union
GCP
Good Clinical Practice
GHTF
Global Harmonization Task Force
GMP
Good Manufacturing Practice
ICH
International Council for Harmonisation of Technical
Requirements for Pharmaceuticals for Human Use
IEC
International Electrotechnical Commission
IFU
Instructions For Use
IMDRF
International Medical Device Regulators Forum
ISO
International Organization for Standardization
MDD
Council Directive 93 / 42 / EEC of 14 June 1993 concerning
medical devices
MDR
Regulation (EU) 2017 / 745 of the European Parliament and
of the Council of 5 April 2017 on medical devices
MAH
Marketing Approval Holder
NMPA
National Medical Products Administration (China)
PTR
Product Technical Requirement
QMS
Quality Management System
SAMR
State Administration for Market Regulation (China)
SFDA
State Food and Drug Administration (China)
UK
United Kingdom

List of Tables
IV
List of Tables
Table 1: Medical Device Classification (Body-contacting devices) ................. 9
Table 2: Medical Device Classification (Non-body-contacting devices) ........ 10
Table 3: Additional information in Chinese label and IFU ............................. 14
Table 4: Application Procedures ................................................................... 16
Table 5: Application timeline ......................................................................... 18
Table 6: Documentation for initial registration ............................................... 19
Table 7: Documentation for change application ............................................ 20
Table 8: Documentation for renewal application ........................................... 20
Table 9: Comparison between “agent” and “authorised representative” ....... 23
Table 10: Criteria of equivalency .................................................................. 29
Table 11: Discrepancies of requirements on the EC ..................................... 34
Table 12: Comparison of requirement on clinical evaluation......................... 35
Table 13: Comparison of requirements on submission dossier .................... 37
Table 14: Requirements on registration testing ............................................ 38
Table 15: Non-conformities to Chinese regulations ...................................... 42
Table 16: Requirement on marketing approval from the country of origin ..... 43
Table 17: List of important Chinese regulations ............................................ 46
Table 18: List of useful Chinese guidelines ................................................... 47
List of Figures
Figure 1: Marketing approval procedure ....................................................... 17

1. Background
5
1. Background
With its large population, the People's Republic of China (will be abbreviated
as “China” in the rest of this thesis) is not an ignorable market to most foreign
medical device manufacturers, many of which are based in the EU. However,
having a marketing approval in the EU does not guarantee a marketing
approval in China. To many European manufacturers, the Chinese medical
device regulations are complex, challenging and ever-changing.
In March 2013 the former State Food and Drug Administration (SFDA) has
been reconstructed into the China Food and Drug Administration (CFDA),
subsequently the Chinese medical device regulations went through many
changes and the CFDA attempted to better normalize the review and
approval procedures for medical device applications. Since the new version
of the "Regulations for the Supervision and Administration of Medical
Devices"
[1]
and the "Provisions for Medical Device Registration"
[2]
came into
force in 2014, more new regulations and guidelines have been issued in
China, providing clarifications to critical issues such as the requirement on
clinical evaluations. Meanwhile, China actively participated in international
exchanges of medical device regulations. In 2013 China has become a full
member of the IMDRF
[3]
and in 2017 China has become a regular member of
the ICH
[4]
. As the approaches to reform the medical device approval system
in China, the CFDA issued the “Special Review Procedure of Innovative
Medical Devices (interim version)” in 2014
[5]
and in January 2018 finalized
the “Guideline on Accepting Foreign Clinical Data” within just 3 months after
the draft being published
[6] [7]
. Compared to the previously medical device
approval system that mainly based on local test data, such approaches
indicated positive signs that China is being more open to new medical
technologies. Additionally, many significant changes have been proposed in
the "Regulations for the Supervision and Administration of Medical Devices
(draft version)"
[8]
that have been submitted to the state council in November
2017, the proposals are currently under discussion.
In March 2018 the CFDA has been reconstructed into the National Medical
Products Administration (NMPA), which is now under the supervision of the
State Administration for Market Regulation (SAMR). New changes to the
medical devices regulations in China are to be expected. In August 2018 the

1. Background
6
SAMR issued the “Provisions for Adverse Events Surveillance and Re-
assessment of Medical Devices”
[9]
. The post market surveillance of medical
devices in China will be strengthened. In November 2018 the NMPA revised
and finalized the “Special Review Procedure of Innovative Medical Devices”
[10]
. The qualification of innovative devices has been further clarified and the
review procedure will be normalized.
1.1 Aims and objectives
The aim of this thesis is to provide a general but systematic review of the
current medical device regulations in China and analyse the gap between the
regulatory requirements in China and in the EU, with the focus on application
procedures for imported medical devices in China and the consideration of
possible challenges to be faced by European medical device manufacturers
that comply already with the medical devices regulations in the EU.
This thesis will also analyse the new trend in Chinese medical device
regulations and discuss the possible opportunities and challenges to be
brought to European medical device manufacturers.
1.2 Notes
Considering that many regulations (e.g., the classification system and
technical documentation requirements) of in vitro diagnostic devices are
significantly different from those of medical devices both in China and in the
EU, this thesis will not cover in vitro diagnostic devices.
Most of the Chinese regulations have been published in Chinese only. Some
of the articles and requirements quoted in this thesis have been translated
from the original Chinese texts into English by the author of this thesis. Notes
will be added when the quoted texts are translated from the original Chinese
texts.

2. Marketing Approval System for Medical Devices in China
7
2. Marketing Approval System for Medical Devices in
China
According to Article 2 and Article 5 of the “Provisions for Medical Device
Registration”, all medical devices sold and used in China shall apply for
registration or filing. Class I medical devices shall apply for filing (refer to
chapter 3.3); class II and class III medical devices shall apply for registration
(refer to chapter 3.2). For imported medical devices, the applications should
be submitted to the NMPA. A foreign applicant shall appoint an agent in
China to conduct the communication with the NMPA and activities defined by
corresponding regulations. According to Article 12 of the same “Provisions”,
the language of submission dossiers should be Chinese; regarding original
documents of foreign languages, both the original copy and translated
Chinese copy should be submitted.
2.1 Qualification of the applicant
As regulated by Article 6 and Article 9 of the “Provisions for Medical Device
Registration”, the registration applicant or filing entity brings the products to
the market in his own name and shall establish and maintain the quality
management system that covers the design and manufacture of the products
to be approved or filed in China. This requirement basically restricted the
registration applicant / filing entity to the “manufacturer” as defined by the
MDD and the MDR.
2.2 “Agent” of Imported Medical Devices
In China, the role of “Agent” of imported medical devices is similar to the role
of “Authorised Representative” in the EU. Both shall be responsible for the
communication between the manufacturer and the authority, and for post
market surveillance activities according to the corresponding regulations. The
name and address of the agent shall appear on the Chinese label and in the
Chinese IFU of imported medical devices in China, while the name and
address of the authorised representative shall appear on the label and in the
IFU of medical devices to be marketed in the EU.
Neither the MDD nor the AIMDD include detailed description of the role and
obligations of an authorised representative, giving the flexibility to define the

2. Marketing Approval System for Medical Devices in China
8
detailed responsibilities in the contract between the manufacturer and the
authorised representative. However more detailed requirements have been
included in the MDR. In China, even though the “Provisions for Medical
Device Registration” has already defined certain obligations of an agent, the
NMPA published a draft version of the “Provisions for the Supervision and
Administration of Agents of Imported Medical Devices”
[11]
in August 2018,
giving more detailed definition of the role and obligations of an agent.
According to this draft, the agent of imported medical devices shall have the
following obligations (note: translated into English by the author of this
master thesis):
Apply for filing and registration according to corresponding regulations;
Monitor and report adverse events from devices sold in China;
cooperate in event investigations;
Conduct product recalls and report to authorities;
Assist the authorities in investigating violations of Chinese regulations
by foreign “Marketing Approval Holders” (MAHs); (note: new
requirement)
Cooperate in product sampling and evaluation of product quality and
provide information and data as requested by the authorities; (note:
new requirement)
Maintain the traceability of devices sold in China and keep the
information of domestic marketing and distributions; (note: new
requirement)
Communicate with authorities and foreign MAHs and inform the latter
about relevant regulations and technical requirements (also amended
ones) in timely manner;
Assist foreign MAHs in completing the remaining tasks from
conditional approvals
1
; (note: new requirement)
Take care of domestic complaints and inform foreign MAHs about
related information; (note: new requirement)
Undertake joint responsibility for product quality and violation of
regulations.
1
Translated into “conditional approvals” by the author, because such certificates regularly
contain the wording “approved based on the following conditions …“.

2. Marketing Approval System for Medical Devices in China
9
2.3 Classification of Medical Devices
As defined by Table 1 and Table 2 below from the “Rules for Classification of
Medical Devices”
[12]
issued by the CFDA in 2015, medical devices are
classified into class I, II and III based on their risk levels.
Body-contacting device
Non-
active
device
Status of use
Patterns of use
Temporary use
(< 24 h)
Short-term use
(≥ 24 h; < 30 day)
Long-term use
(≥ 30 day)
Skin
/ Orifice
(openings)
Trauma
/
Tissue
Blood
circulation
/ Central
nerve
Skin
/ Orifice
(openings)
Trauma
/
Tissue
Blood
circulation
/ Central
nerve
Skin
/ Orifice
(openings)
Trauma
/
Tissue
Blood
circulation
/ Central
nerve
1
Liquid
transportation
device
Ⅱ
Ⅱ
Ⅲ
Ⅱ
Ⅱ
Ⅲ
Ⅱ
Ⅲ
Ⅲ
2
Blood and other
body fluids
alternation
device
-
-
Ⅲ
-
-
Ⅲ
-
-
Ⅲ
3
Medical
dressing
Ⅰ
Ⅱ
Ⅱ
Ⅰ
Ⅱ
Ⅱ
-
Ⅲ
Ⅲ
4
Invasive device
Ⅰ
Ⅱ
Ⅲ
Ⅱ
Ⅱ
Ⅲ
-
-
-
5
Reusable
surgical device
Ⅰ
Ⅰ
Ⅱ
-
-
-
-
-
-
6
Implantable
device
-
-
-
-
-
-
Ⅲ
Ⅲ
Ⅲ
7
Contraceptive
and family
planning device
(excluding
reusable
surgical device)
Ⅱ
Ⅱ
Ⅲ
Ⅱ
Ⅲ
Ⅲ
Ⅲ
Ⅲ
Ⅲ
8
Other non-
active devices
Ⅰ
Ⅱ
Ⅲ
Ⅱ
Ⅱ
Ⅲ
Ⅱ
Ⅲ
Ⅲ
Active
Device
Status of use
Patterns of use
Minor injury
Moderate injury
Serious injury
1
Energy
treatment
device
Ⅱ
Ⅱ
Ⅲ
2
Diagnostic and
monitoring
device
Ⅱ
Ⅱ
Ⅲ
3
Liquid
transportation
device
Ⅱ
Ⅱ
Ⅲ
4
Ionizing
radiation
device
Ⅱ
Ⅱ
Ⅲ
5
Implantable
device
Ⅲ
Ⅲ
Ⅲ
6
Other active
devices
Ⅱ
Ⅱ
Ⅲ
Notes: 1. “I”, “II” and “III” herein respectively refer to class I, II and III medical devices.
2. “-” herein means the situation is inapplicable to any class of medical devices.
Table 1: Medical Device Classification (Body-contacting devices)

2. Marketing Approval System for Medical Devices in China
10
Non-body-contacting device
Non-
active
device
Status of use
Patterns of use
little impact
Minor impact
Significant impact
1
Nursing device
Ⅰ
Ⅱ
-
2
Device for medical
device sterilization
and cleaning
-
Ⅱ
Ⅲ
3
Other non-active
devices
Ⅰ
Ⅱ
Ⅲ
Active
device
Status of use
Patterns of use
little impact
Minor impact
Significant impact
1
Clinical laboratory
instruments
Ⅰ
Ⅱ
Ⅲ
2
Stand-alone
software
-
Ⅱ
Ⅲ
3
Instruments for
medical devices
disinfection and
sterilization
-
Ⅱ
Ⅲ
4
Other active devices
Ⅰ
Ⅱ
Ⅲ
Notes: 1. “I”, “II” and “III” herein respectively refer to class I, II and III medical devices.
2. “-” herein means the situation is inapplicable to any class of medical devices.
Table 2: Medical Device Classification (Non-body-contacting devices)
However while in the EU medical devices are to be classified based on
certain general rules set out in Annex IX of the MDD or Annex VIII of the
MDR, in China the classification of a medical device has to be made in
accordance with the “Medical Device Classification Catalogue”
[13]
and a
product code shall be assigned to each medical device accordingly. The
above-mentioned catalogue will be updated from time to time and the current
version has been issued in 2017 by the CFDA. Compared to the previous
version, this new catalogue has expanded from 22 sub-catalogues to 43 sub-
catalogues and examples have been increased from 1008 to 6609, with the
addition of indication and description for each medical device and the
reclassification of medical devices from 40 categories into the lower class.
[14]
Finding the correct product code not only determines the product
classification but also helps identifying the applicable Chinese standards
correctly. In case there is no applicable category for the device to be
classified, the applicant can apply for the correct classification or submit
directly to the NMPA as a class III device (the NMPA will determine the
correct classification during the technical review).

2. Marketing Approval System for Medical Devices in China
11
2.4 Registration Units
It is very common that manufacturers combine different medical devices or
components for the clinical application as systems or procedure packs.
According to Article 12 of the MDD and Article 22 of the MDR, no extra CE
marking will be needed for systems or procedure packs if the requirements
have been met. This actually gives the flexibility in choosing different CE
marking approaches for the combination of medical devices or components,
either treating the combination as a device in its own right or applying for
conformity assessment for each component included individually.
In China, the applicant has less flexibility in defining a registration unit. In
2017 CFDA issued the “Guideline on Division of Registration Unit”
[15]
,
providing clarifications on the criteria for dividing different products and
components into separated applications. Incorrect defined registration unit
could result in the submission being rejected. According to the above-
mentioned guideline, the following rules should be considered (note:
translated into English by the author of this master thesis):
a) Active devices
Devices of different technical principle should be submitted separately;
Devices of different structure / component that affects product safety
and efficacy should be submitted separately;
Devices of different performance criteria that leads to different
application scope or mechanism should be submitted separately;
Devices of substantially different application scope should be
submitted separately;
Active devices and the non-active consumables that have to be used
together should be submitted separately;
Independent active devices to be used together in the same
application scope should be submitted separately;
The main console and its applied parts should be submitted in the
same submission;
Accessories with different intended use should be submitted
separately;

2. Marketing Approval System for Medical Devices in China
12
Active accessories and non-active accessories that are regulated as
medical devices should be submitted separately unless they are
packed in the same sterile packaging.
b) Non-active devices
Devices of different technical principles should be submitted
separately;
Devices with and without drug / active ingredients should be submitted
separately;
Devices of different surface treatments or structures that affect the
safety and efficacy should be submitted separately;
Devices of different physical appearances that affect the safety and
efficacy should be submitted separately;
Non-active devices and its active accessories should be submitted
separately;
Devices of different structures/components or processed by different
procedures that affect the safety and efficacy should be submitted
separately;
Single use and reusable devices should be submitted separately if the
difference affects the performance criteria;
Devices treated by different sterilization processes should be
submitted separately if the difference affects the performance criteria;
Devices of different structure that leads to different performance
criteria or different application scope should be submitted separately;
Devices to be used in combination with different products that affects
the performance criteria should be submitted separately;
Devices of different animal origins should be submitted separately;
Devices of different application scopes should be submitted separately.
2.5 Marketing Approval Certificates
The registration certificates issued by the NMPA to class II and class III
imported medical devices are valid for 5 years and should have the
information listed below:
[16]
(note: translated into English by the author of this
master thesis)
Certificate No.
Name of the applicant

2. Marketing Approval System for Medical Devices in China
13
Registered address of the applicant
Address of the manufacturing site
Name of the agent
Registered address of the agent
Product name
Models and specifications
Structure and components
Application scope
Attachment: Product Technical Requirement (PTR)
Notes (in case of conditional approval)
Issue date and expiry date
According to Article 49 of the “Provisions for Medical Device Registration”,
approval is requested for any change to the content on this certificate and its
attachment(s). Special attention should be paid to the attached PTR, which
will include technical specifications, testing criteria and testing methods of the
approved medical device. For each change in the PTR, even not being the
major change, a change application should be submitted. The importance of
PTR will also be discussed in later chapters of this thesis.
A change approval is a supplement to the initial certificate and is valid until
the expiry date of the initial certificate. It should have the information listed
below:
[16]
(note: translated into English by the author of this master thesis)
Certificate No. (of initial certificate)
Product name
Change description (before and after change)
Notes: this document is to be used with “xxxx” certificate
Approval date
For class I medical devices, only an acceptance notice of the filing will be
issued, which has no expiry date.
[16]
2.6 Label and IFU
As regulated by the “Provisions for Instructions and Labels of Medical
Devices”
[17]
issued by the CFDA in 2014, the medical devices being sold and

2. Marketing Approval System for Medical Devices in China
14
used in China should be accompanied by the Chinese label and IFU.
However, the Chinese label and IFU should not be a simple translation of the
label and IFU used in the EU. Additional information has been requested by
the provisions mentioned above, in particular the following:
Additional information
Chinese
label
Chinese
IFU
Manufacturing site address (if applicable, the name and
address of contract manufacturer)
x
x
Name, address and contact information of the agent
x
x
Registration certificate number or filing number
x
x
PTR number
x
Manufacturing date, shelf life or expiry date
x
x*
*Note: this is a mandatory section of the Chinese IFU; wordings like “please refer to the
label” should be included when it’s not practical to include the manufacturing date / expiry
date in the IFU.
Table 3: Additional information in Chinese label and IFU

3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
15
3. Application Procedures for Marketing Approvals of
Imported Medical Devices in China
3.1 Application Types
For class I medical devices, filings should be submitted for initial record or for
changes of a filed record. No technical review will be involved.
[2]
For class II and class III medical devices to be marketed in China for the first
time, initial registration application should be submitted, and the approval will
be issued after the technical review for initial registration. For changes of
administrative information on the registration certificate or its attachments, a
filing should be submitted, and no technical review will be involved; for
changes to other approved items on the registration certificate or its
attachments, a change application should be submitted, and the approval will
be issued after the technical review for change application.
A renewal application should be submitted at least 6 months before the
expiry date of the certificate and the approval will be issued after the
technical review for renewal application.
[2]
For changes to approved IFUs of class II and class III medical devices, filings
should be submitted according to a specific IFU change procedure. A
rejection notice will be issued if the change should involve technical review,
in which case a change application should be submitted instead. If no
rejection notice has been issued within 20 working days after the acceptance
date, then the proposed IFU changes can be implemented.
[18]
For imported medical devices, all the above-mentioned filings or applications
should be submitted to the NMPA.
[2]
3.2 Application procedures for approval of class II and class III
imported medical devices
The CMDE website listed the following procedures that the applicant has to
take into consideration during the preparation of the application, the technical
review by the CMDE and the post market phase for class II and class III
imported devices.
[19]

3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
16
Mandatory procedure
Optional procedure
Preparation phase
Pre-application consulting
Classification application
Type testing
Clinical evaluation
Innovation qualification
Technical review
phase
Acceptance of application
Priority qualification
Supplement request
Expert meeting
Rejection / Withdrawal
Approval
Post market phase
Change application
Renewal application
IFU change application
Table 4: Application Procedures
Type testing and clinical evaluation are both categorized as mandatory
procedure for applications in China, while in the EU type testing is not
mandatory but depends on which conformity assessment route the
manufacturer selects. The detailed requirements on type testing and clinical
evaluation will be discussed in chapter 4 of this thesis.
3.2.1 Approval procedures (flowchart)
According to the guidelines issued by the CFDA in 2017 on the initial
registration, the change application and the renewal application for class II
and class III imported medical devices
[20] [21] [22]
, the processes are similar
and can be summarized into the flowchart from Figure 1 below:
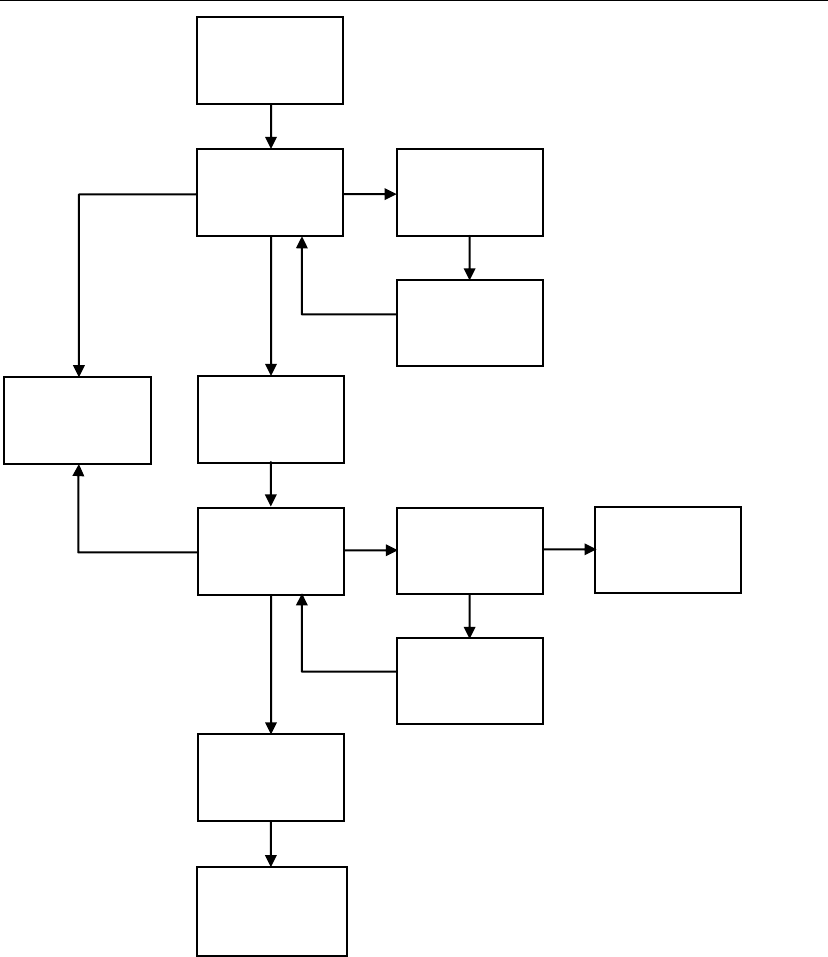
3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
17
Figure 1: Marketing approval procedure
Notes:
1. Formal inspection to be conducted by the administrative service centre of the NMPA;
2. Decision of acceptance to be made by the administrative service centre of the NMPA;
3. Technical review to be conducted by the CMDE;
4. Only 1 chance of supplement submission (must be submitted within 1 year); only 3
face-to-face discussions with the reviewer will be allowed; only 1 chance of pre-
review before the supplement submission will be allowed.
3.2.2 Application timeline
The timelines for initial registration, change application and renewal
application in China can be summarized into Table 5 below:
Certificate
issued
Formal
inspection
1
Application
submitted
Application
accepted
2
Application
approved
Technical
review
3
Application
rejected
Application
re-submitted
1
Corrective
notice
1
Supplement
submission
4
Supplement
notice
4
Application
withdrawn

3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
18
Procedure
Timeline
Formal inspection
5 working days
Transfer of documentation
3 working days
Technical review
Class II: 60 working days
Class III: 90 working days
Decision of approval
20 working days (+ 10 working days if necessary)
Issue of certificate
5 working days
Notes:
1. Clock stops on the issue date of supplement notice and re-starts on the reception
date of supplement submission
2. The above listed timeline does not include extra time for expert meetings and / or on-
site audit
Table 5: Application timeline
3.2.3 Application requirements
3.2.3.1 Initial registration of imported medical devices
For class II and class III imported medical devices to be marketed in China
for the first time, a dossier including the following sections listed in Table 6
below should be submitted for initial registration applications according to the
“Guideline on Initial Registration of Imported Medical Devices” issued by the
NMPA in 2018.
First level title
Second level title
1. Application form
2. Qualification files
3. Basic requirement list for the
safety and efficacy
4. Summary info
4.1 Overview
4.2 Product description
4.3 Models and specifications
4.4 Packaging description
4.5 Intended use and contraindications
4.6 Equivalent product or product of
previous generation (if applicable)
4.7 Others
5. Research data
5.1 Performance
5.2 Biocompatibility
5.3 Biological safety

3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
19
5.4 Disinfection and sterilization process
5.5 Shelf life and packaging
5.6 Animal studies
5.7 Software
5.8 Others
6. Manufacturing info
6.1 Manufacturing process of non-active /
active devices
6.2 Manufacturing sites
7. Clinical evaluation
8. Product risk assessment
9. Product technical requirement
10. Registration test report
10.1 Test report
10.2 Pre-evaluation conclusion
11. IFU and labels
11.1 IFU
11.2 Label samples of units for sale
12. Declaration of conformity
Note: translated into English by the author of this master thesis.
Table 6: Documentation for initial registration
3.2.3.2 Change application
As regulated by Article 49 of the “Provisions for Medical Device Registration”,
in China, applications should be submitted for all changes to the initial
registration certificate and related attachments. For changes to administrative
information, a filing should be submitted; for other changes, a change
application for approval should be submitted.
According to the “Guideline on Change Application of Imported Medical
Devices”
issued by the NMPA in 2018, a dossier including the following
sections listed in Table 7 below should be submitted for change applications
of class II and class III imported medical devices.
Section
Notes
1. Application form
2. Qualification files
3. Declaration on changes
4. Copy of initial approval
certificate and previous change
approvals
5. Descriptions of changes
Change comparison form and explanations

3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
20
on the following items, if applicable:
- product name;
- product technical requirement;
- model and specifications;
- structure and components;
- intended use;
- on manufacturing site address;
- on other content in the certificate;
- on other changes
6. Risk management report
regarding the changes
7. Evaluation on changes to the
safety and efficacy
*Clinical evaluation is a must for changes
to intended use.
8. Registration test report
regarding changes to the PTR
9. Declaration of conformity
- Conformity to Chinese regulations and
standards, with a list of applicable
Chinese standards;
- Self-declaration on truthfulness
Note: translated into English by the author of this master thesis.
Table 7: Documentation for change application
3.2.3.3 Renewal application
According to the “Guideline on Renewal Application of Imported Medical
Devices”
[22]
issued by the NMPA in 2018, a dossier including the following
sections listed in Table 8 below should be submitted for the renewal of
existing marketing approval of class II and class III imported medical devices.
Section
Notes
1. Application form
2. Qualification files
3. Declaration on no change
*no change to the most recent
approval (initial or change
approval)
4. Copy of initial approval certificate and
previous change approvals
5. Summary and related documentation on
the remaining tasks required by
conditional approval.
*the previously requested
summary report on post
marketing surveillance has been
exempt in the 2018 revision
issued by NMPA
[18]
Note: translated into English by the author of this master thesis.
Table 8: Documentation for renewal application

3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
21
Since the issue of the “Provisions for Medical Device Registration” in 2014,
the CFDA offered to simplify the procedure of renewal application and
accelerate the technical review. To achieve such simplification and
acceleration, the renewal application has been restricted to the “non-change
renewal application” and has to be submitted separately from change
applications
[16] [22]
. The previous approach of combining the renewal and
change applications together are no longer allowed. In 2018 the NMPA
revised again the documentation requirement for renewal applications
[27]
,
further simplifying the documentation for renewal submissions. This is to
align with the new “Provisions for Adverse Events Surveillance and Re-
assessment of Medical Devices”, which requires the MAH to submit
periodically a summary of adverse events, surveillance data, risk assessment
and management to the responsible authorities.
Although the documentation for renewal submission has been simplified, it
doesn’t mean technical review will be omitted. According to Article 55 of the
“Provisions for Medical Device Registration”, the renewal application shall not
be approved if the most recent version of applicable Chinese standards
cannot be met or the tasks listed on the initial registration certificate have not
been completed. Hence it is still possible that additional tests and data being
requested to get the initial certificate renewed. The applicant should review
the up-to-date status of the Chinese standards being referred to and identify
if any new Chinese standards not being referred to could be applicable as
well.
3.3 Filing of class I imported medical devices
The filing of class I imported medical devices should be submitted to the
NMPA. The administrative service centre of the NMPA will check the format
and completeness of the filing dossier. If it complies with the required format,
a notification on acceptation of the filing dossier will be issued upon reception.
According to Article 9 of the current “Regulations for the Supervision and
Administration of Medical Devices”, the following documentation should be
submitted for the filing of class I medical devices (note: translated into
English by the author of this master thesis):
risk assessment documentation;

3. Application Procedures for Marketing Approvals of Imported Medical Devices in China
22
product technical requirement;
product test report;
clinical evaluation documentation;
product IFU and label sample;
quality system documentation related to product development and
manufacture;
other data to demonstrate the product safety and efficacy.

4. Discussion
23
4. Discussion
4.1 Possible challenges from the current medical device regulations in
China
4.1.1 The importance of “Agent”
Chapter 2.2 introduced the role of “Agent” in China and its obligations. To
further discuss the similarity and differences between the role of “Agent” in
China and the role of “Authorised Representative” in the EU, Table 9 below
summarizes the major obligations of these two roles as defined by
corresponding regulations. It is worth noting that both in China and in the EU
the trend is to better supervise the local agent / representative.
Agent in China
Authorised
Representative in the EU
Role and obligations
Current
provisions
Planned
provision
(draft)
MDD /
AIMDD
MDR
The sole representative
x
x
Communication between
the authority and the
manufacturer
x
xx
x
xx
Establish a quality
management system
x
xx
Submit the application
for filing or registration
x
x
Provide to the authority
the evidences of
regulatory compliance
x
x
x
xx
Post market surveillance
and vigilance activities
x
xx
x
xx
Note: x = covered; xx = strengthened
Table 9: Comparison between “agent” and “authorised representative”
The contract between the agent and the foreign applicant is not the subject of
review by the NMPA. It is the responsibility of both the applicant and the
agent to make sure that the contract covers the requested obligations
completely. In each application the agent shall submit to the NMPA a
declaration on fulfilling all the obligations defined by regulations in China.

4. Discussion
24
4.1.2 Possible classification differences
The medical device classification rules in China have been explained in
chapter 2.3. The most significant difference between the classification rules is
that in China the intended duration of use has been divided into “temporary”
(< 24 h), “short-term” (≥ 24 h; < 30 day) and “long-term” (≥ 30 day), while
both the MDD and MDR divide the duration into “transient” (< 60 min), “short-
term” (≥ 60 min; ≤ 30 day) and “long-term” (> 30 day). Due to this difference,
the “short-term” devices in the EU that claim to be used for at least 60
minutes but less than 24 hours will be defined as “temporary” devices in
China and possibly classified into a lower class than in the EU.
Certain special rules from the same classification rules mentioned above will
also cause discrepancies, such as:
A medical device supplied in sterile state shall always be classified as
class II or above;
A medical device with measuring function shall always be classified as
class II or above.
Because technical review by the CMDE is mandatory for class II and class III
imported medical devices, if a class I device in the EU has been classified
into higher class in China, it is possible that the CMDE will review extra data
and information that have never been requested by the notified bodies.
4.1.3 Possible difference in registration unit
According to the rules described in chapter 2.4 for defining a registration unit
for medical device submissions in China, it is possible that the compilation of
a system / procedure pack (the registration unit) in China has to be defined
differently from the approved system / procedure pack in the EU. According
to Article 12 of the MDD, the entity that puts the system / procedure pack on
the European market does not have to be the “manufacturer” of the system /
procedure pack. However as explained already in chapter 2.1, in China the
registration applicant or filing entity has to be the manufacturer of the
registration unit. Hence it will cause issues if the applicant does not have the
quality management system that covers the design and manufacture of the
whole system / procedure pack (registration unit) to be approved or filed in
China.

4. Discussion
25
4.1.4 Possible issues with the Chinese label and IFU
Chapter 2.6 summarized the additional requirements on the Chinese label
and IFU. It is worth noting that according to section 13.1 in Annex I of the
MDD and section 23.1 (d) in Chapter III of the MDR, instructions for use shall
not be required for class I and class IIa devices if such devices can be used
safely without any such instructions. However, in China, no such exemption
has been included in the provisions mentioned above.
It is also worth noting that although the Chinese label and IFU of medical
devices are also subject to review by the NMPA, they are not issued as
attachments of the approval certificate for medical devices. According to
Article 49 of the “Provisions for Medical Device Registration”, only changes to
the certificate and related attachments are subject of review and approval by
the NMPA. For changes to the approved Chinese IFU of class II and class III
devices, according to the “Filing Procedure of Changes to Medical Device
IFUs”
[18]
published in 2015, a filling should be submitted to the NMPA, as
explained in chapter 3.1.
4.1.5 Triggers for an “initial registration”
Chapter 3.2.3.1 deals with the documentation to be submitted for an “initially
registration” of class II and class III medical devices in China. Such an “initial
registration” literally applies to any device that has never been marketed in
China before. However, based on the experiences from the author of this
thesis, the NMPA may request the applicant to submit an “initial registration”
if the proposed changes of the approved medical device are considered by
the NMPA as significant. Additionally, if the renewal application for the
existing certificate has not been submitted and accepted by the NMPA within
the regulated deadline, an “initial registration” shall be submitted instead.
Chapter 4.1.6.2 will further discuss when a change application could trigger
an initial registration submission.
4.1.6 Possible issues with the submission dossier
4.1.6.1 Initial registration
The general structure and requirements of the initial registration submission
dossier of class II and class III medical devices in China share a lot of

4. Discussion
26
common features with the GHTF document “Summary Technical
Documentation for Demonstrating Conformity to the Essential Principles of
Safety and Performance of Medical Devices (STED)”
[23]
. Considering that the
technical documentation in accordance with the MDD or the MDR also
shares a lot of common features with the STED, the existing data generated
for the technical documentation for CE approval should be able to cover a
major part of the submission dossier for initial applications in China.
Special attention should be paid to the following sections, which would
require additional data in comparison to the technical documentation of a CE
marked device or otherwise cause discrepancies:
A) Product Technical Requirement (PTR)
As requested by Article 15 of the “Provisions for Medical Device
Registration”, for class II and class III medical devices, the PTR should
be submitted for approval, and the devices being marketed in China
should comply with the approved PTR.
According to the “Guideline on drafting Product Technical
Requirement”
[24]
issued by the CFDA, the PTR should include the
following content: (note: translated into English by the author of this
master thesis)
1) Product name;
2) Product models / specifications and descriptions of all the
differences between them;
3) Performance criteria related to the function, safety and quality
control of the finished product; the criteria should at least meet the
requirements from applicable mandatory Chinese national and
industrial standards.
4) Testing method to verify that the finished product can meet the
criteria.
The PTR is a China specific document. It includes crucial information
from product design as well as necessary quality control measures. It
is crucial for medical device approvals in China not only as an
important attachment to the registration certificate that defines the
product models / specifications and performance criteria, but also as

4. Discussion
27
the link between the product design, quality control and Chinese
standards.
It is worth noting that the Chinese standards will be updated
periodically, and the PTR should always take reference to the latest
version of Chinese standards. It is actually similar to the reference of
latest harmonized standards in the technical documentation for CE
approval. Although many Chinese standards are actually adopted from
harmonized standards such as ISO and IEC standards, discrepancies
and different interpretations do exist. It is also very common that the
most recent Chinese standard is adopted from an obsolete version of
the equivalent harmonized standard because the adoption will take
time. Hence when referring to Chinese standards in the PTR, it is
necessary to evaluate the possible discrepancies between the
requirements in China and in the EU.
B) Registration test report
Chinese test institutions conduct type testing for different purposes.
Type testing for registration purpose is conducted in accordance with
the PTR proposed by the applicant and reviewed by the test institution.
It has to be a full test with test samples manufactured in the controlled
quality management system.
In the EU, type testing is not always necessary to get market
approvals depending on the conformity assessment route selected by
the manufacturer. However, in China, the registration test report
issued by a qualified Chinese test institution is mandatory and the pre-
evaluation conclusion from the same test institution has to be
submitted as well for initial applications according to the current
“Provisions for Medical Device Registration”.
C) Clinical evaluation, clinical trial
According to Article 22 of the current “Provisions for Medical Device
Registration”, clinical trial has to be conducted for all class II and class
III medical devices, unless the product is on the exemption list or it
applies one of the following criteria:

4. Discussion
28
1) Where the functional mechanism of the device is definite, the
design is finalized, the production process is well-established, and
an equivalent marketed medical device has been in clinical use for
years and no serious adverse events are recorded, and its
conventional purposes of use are not changed;
2) Where safety and effectiveness of the device can be proved
through non-clinical evaluation;
3) Where safety and effectiveness of the medical device can be
demonstrated through the analysis and assessment made on the
basis of the data obtained from clinical trial or application of an
equivalent medical device.
Even though the above listed alternative approaches have been
allowed, the criteria of equivalency have not been clarified. In April
2015 the CMDE issued the “Guideline on the Clinical Evaluation of
Medical Devices”
[25]
, which for the first time elucidated the criteria of
equivalency and the requirements on different approaches for clinical
evaluation. In summary, three different acceptable approaches have
been explained in this guideline:
1) Exempted device – the (imported) product is equivalent to an
approved device in China of the same exempted category;
2) Non-exempted device – but (imported) product is equivalent to an
approved device in China;
3) Clinical trial.
The equivalency on items listed in Table 10 below should be taken
into consideration.
Equivalency on
Equivalency to
exempted
device
non-exempted device
(non-active device)
non-exempted device
(active device)
Working principle
x
X
x
Structure
x
X
x
Manufacturing
procedure
X
x
Production
materials
x
(especially
human
contacting
materials)
x
(including material
brand, animal source
material, allogeneic
material, components,
drug components,
biologically active
x
(including material
brand, animal source
material, allogeneic
material, components,
drug components,
biologically active

4. Discussion
29
material, applicable
standards)
material, applicable
standards)
Performance
requirements
X
x
x
(performance
parameters; function
parameters)
Safety evaluation
x
(including
biocompatibility,
biological safety, etc.)
x
(including
biocompatibility,
biological safety, EMC,
etc.)
Core function of
software
x
Applicable
national /
industrial
standards
x
x
Intended use
X
x
(including intended
population, application
areas, human contact
type, indications,
disease stage and
extend of indications,
application
environment)
x
(including intended
population, application
areas, human contact
type, indications,
disease stage and
extend of indications,
application
environment)
Method/Type of
application
X
x
x
Contraindication
x
x
Caution and
warnings
x
x
Sterilization
X
x
x
Packaging
x
x
Labelling
x
x
Instructions for
Use
x
x
Delivery status
x
x
Table 10: Criteria of equivalency
As the table above shows already, to demonstrate the equivalency,
the applicant has to provide for both the reference and new device the
detailed information that is not publicly available, such as production
materials, manufacturing procedure and applicable standards. In
addition, when choosing the equivalency approach, a clinical
evaluation report (CER) has to be provided as the alternative

4. Discussion
30
document to the clinical trial report. The CER should also include the
following, not publicly available information (note: translated into
English by the author of this master thesis):
1) Non-clinical research data;
2) Literatures;
3) Clinical application experience data;
4) Complaints and adverse events;
5) CAPAs related to clinical risks;
6) Data from Chinese population;
7) Additional clinical study in China.
Due to restrictions from the availability of requested information and
data from reference devices, it’s not practical to choose the
equivalency route when only similar products from competitors have
been approved in China. It makes more sense to choose this
approach when the same applicant has similar products or same
products of previous generation that have been approved in China
already. However, the significance of differences still has to be
determined during the case-by-case review. In case of insufficient data,
it would be very challenging to finish the additional clinical study within
the 1-year deadline for supplement submission as restricted by Article
35 of the “Provisions for Medical Device Registration”. The worst case
could be withdrawal of the application due to insufficient data.
If the equivalency approach is not feasible, then clinical trial has to be
conducted for class II and class III medical devices. However, in the
EU, if the device is neither implantable nor in Class III, clinical trial is
not always the first option to demonstrate its safety and efficacy.
According to Article 23 of the “Provisions for Medical Device
Registration”, the clinical trial institution should be qualified in
accordance with the Chinese “Good Clinical Practice”, meaning only
clinical trial data from qualified Chinese institutions would be accepted.
In 2018 the CFDA issued the final version of the “Guideline on
Accepting Foreign Clinical Data”, offering to simplify the application
procedure for applicants with qualified non-local clinical trial data.

4. Discussion
31
Chapter 4.2.1.1 of this thesis will further discuss the meaning and
impact of this guideline.
4.1.6.2 Change application
Chapter 3.2.3.2 introduced the requirements for change applications for class
II and class III medical devices in China. Although no explanation has been
given in the “Provisions for Medical Device Registration”, not all changes can
be approved through change applications. The “Guideline on Division of
Registration Unit”
should be taken into consideration when deciding whether
the proposed changes may trigger a new application (= initial registration) in
China. The key criteria will be the significance of differences between the
technical principle, structure / component, performance criteria and
application scope. However, whether the changes are significant enough to
trigger a new application, interpretations from the applicant, the
administrative service centre of the NMPA and different reviewers from the
CMDE could be different. It is possible that an accepted change application
being rejected during the technical review and a new application for the
changed product has to be submitted instead.
In the EU, the manufacturer can always consult the notified body on the
necessity of a change notification or the application for a new certificate. In
China however, although the NMPA offers pre-submission consultation to
applicants, the communication will be much more limited than that between
the manufacturer and the notified body in the EU. As explained in the CFDA
announcement on the implementation of pre-submission consultation
[26]
, only
very few appointments will be scheduled, and the applicant has to apply for
an appointment far in advance. Also, the duration of consultation will be very
short and only up to 5 questions are allowed to be posted by an applicant in
each consultation. Furthermore, different offices from the CMDE will attend
the consultation in rotation and only questions related to the responsibilities
of the attending office can be asked during the consultation.
Considering the
uncertainties, it is recommendable to conduct the case-by-case assessment
of the impact from proposed changes to existing approvals in China.
Regarding the dossier requirement, special attention should still be paid to
the possible involvement of registration test and clinical evaluation. The

4. Discussion
32
necessity of additional tests to be done in China should be evaluated
thoroughly before the submission because it would be very difficult to finish
them later within the 1-year deadline of supplement submission.
4.2 Recent changes indicating the new trend in Chinese medical
device regulations
The publication of the “Provisions for Medical Device Registration”
in 2014
has been followed by the publication of many other new regulations and
revisions to existing regulations in China, some of which indicated very
intriguing new trend. This chapter will discuss the most significant new
changes in Chinese medical device regulations.
4.2.1 Clinical trial exemption
4.2.1.1 The acceptance of foreign clinical trial data
As mentioned already in section 4.1.6.1 of this thesis, clinical trial is a must
for all class II and class III medical devices unless meeting the criteria of
exemption according to the current “Provisions for Medical Device
Registration”. However, it is difficult to get such exemptions, i.e., to obtain all
the requested information to demonstrate the equivalency to devices that
have been approved in China already. Hence the publication of the
“Guideline on Accepting Foreign Clinical Data” is definitely good news to
foreign applicants because due to the qualification of clinical trial institution,
local clinical trial data will be necessary for imported class II and class III
medical devices that cannot be exempted from clinical trials. However, this
new guideline will not put an end to the need of local clinical trial data for
imported class II and class III medical devices, but rather provides an
alternative choice for those applicants with qualified existing data.
First of all, the data covered in this guideline should meet the following
criteria (note: translated into English by the author of this master thesis):
clinical trial data to assess the product safety and efficacy under
normal use condition
obtained from qualified clinical institutions
for the same device that applies for approval in China

4. Discussion
33
Secondly the data should be obtained following the basic requirements listed
below:
in accordance with the Helsinki Declaration;
in accordance with the Chinese “Good Clinical Practice for Medical
Devices”
[28]
(any discrepancy should be justified);
the applicant and clinical trial institution should be the subject of
supervision and inspection from the NMPA;
the data obtained should be truthful, reliable and traceable and the
complete data should be provided to the NMPA.
The data to be submitted to the NMPA should include at least the clinical trial
protocol, conclusion from the ethic committee and complete clinical trial
report with the analysis of the complete data and its conclusion.
If applicable Chinese guidelines on the same or similar product category
include requirements on the clinical trial such as trial design, sample size and
endpoints, the foreign data have to meet these Chinese requirements. And in
case of discrepancies that cannot be justified, additional clinical study in
China will be requested. The differences between factors such as races,
living conditions and medical facilities that may affect the clinical trial result
should also be taken into consideration and should be justified.
4.2.1.2 The challenges and opportunities
While providing an alternative approach to conducting local clinical trials in
China, the data that may be accepted will be restricted to data with good
quality from a full-scale clinical trial. However, in the EU, a full-scale clinical
trial is not always the first option for medical device manufacturers. Hence
this new guideline will not be helpful to manufacturers that have never
conducted a full-scale clinical trial. Additionally, the MDD doesn’t require the
clinical investigations for medical devices to be conducted in accordance with
the GCP. Although the MDR now requires that clinical investigations for
medical devices should be in line with the international standard ISO
14155:2011
( 2 )
on good clinical practice, it should be noted that ISO
14155:2011 does not cover many aspects that have been required by the
(2)
ISO 14155:2011, Clinical investigation of medical devices for human subjects -- Good
clinical practice.

4. Discussion
34
GCP that were primarily developed for clinical trials of medicinal products. It
is also worth noting that although the Chinese GCP takes reference from the
ICH Guideline on Good Clinical Practice
[29]
, there are noticeable
discrepancies aside from Chinese local laws and regulations. As an example,
Table 11 shows the discrepancies of requirements on the ethics committee
(EC) between the Chinese GCP, the ICH Guideline and ISO 14155:2011.
Chinese GCP
ICH Guideline on GCP
ISO
standard
14155:2011
Composition
of the EC
- At least five members,
including members from
medical and non-medical
area and members of
different gender;
- At least one member of
legal occupation among
members from non-
medical area.
- At least one member who
is independent of the
institution / trial site.
(a) At least five members.
(b) At least one member
whose primary area of
interest is in a non-
scientific area.
(c) At least one member who
is independent of the
institution / trial site.
N / A
Document
retention
The EC should retain all
relevant records for a
period of at least 10-years
after completion of the trial.
The EC should retain all
relevant records for a period
of at least 3-years after
completion of the trial.
N / A
Table 11: Discrepancies of requirements on the EC
Despite discrepancies, both China and the EU have been trying to harmonize
the clinical trial requirements with international regulations. And since the
CFDA became the regular member of ICH in 2017, the harmonization
procedure in China has been accelerated. The publication of the “Guideline
on Accepting Foreign Clinical Data” is CFDA’s first attempt to save
unnecessary cost, time and effort for applicants who have already obtained
solid data in compliance with recognized international regulations.
The applicant should carefully review the existing clinical trial data and
analyse possible gaps between the relevant requirements in China and in the
EU. If discrepancies cannot be fully justified, additional clinical study in China
should be taken into consideration. CFDA recommended that the applicant
should consult CFDA’s reviewer first before submitting registration
applications with foreign clinical trial data only. Considering the 1-year
deadline for the supplement submission, it would make sense to do some

4. Discussion
35
pre-preparation works for possible additional clinical study in China before
the submission, such as screening potential clinical study site(s), planning for
budget and resource. In case additional clinical data have been requested
during the review, the applicant should make full use of the very limited
chances to discuss with the reviewer on the design and sample size of the
clinical study and possible extension of the review time.
Now that the MDR also requires higher quality for clinical data, if planned in
advance and covering the specific requirements from Chinese GCP in the
clinical investigation protocol, there could be good chance of using the same
clinical data for both the conformity assessment in the EU and the
registration application in China.
4.2.2 Proposed clinical evaluation exemption for class II medical devices
In November 2017 the "Regulations for the Supervision and Administration of
Medical Devices (draft version)" has been submitted to the state council of
China. This draft version included many proposed revisions to the current
“Regulations for the Supervision and Administration of Medical Devices”.
One
significant revision is to the requirement on clinical evaluation. Table 12
compares the relevant articles in the current version and the proposed
version of the above-mentioned regulations and in the current version of
“Provisions for Medical Device Registration” (note: translated into English by
the author of this master thesis).
"Regulations for the Supervision and Administration
of Medical Devices"
“Provisions for
Medical Device
Registration”
(2014)
Proposed version (2017)
Current version
(2014)
Article 17:
… For application for registration
of a class II medical device,
clinical evaluation is not
mandatory; For application for
registration of a class III medical
device, clinical evaluation is
mandatory …
Article 19:
… For application for
registration of a class
II or class III medical
device, clinical trial
shall be conducted…
Article 22:
… For application for
registration of a class
II or class III medical
device, clinical trial
shall be conducted…
Table 12: Comparison of requirement on clinical evaluation

4. Discussion
36
Due to some of the proposed revisions being very significant, the discussion
on this new "Regulations for the Supervision and Administration of Medical
Devices" is expected to take quite some time and there is no guarantee that
all of the proposed revisions will be approved by the state council. However,
this proposal indicated intriguing signs already. Firstly, clinical trial will no
longer be the first option to apply for a registration certificate in China;
compared to previously restricting to clinical trial only, the NMPA is now
trying to accept alternative clinical evaluation approaches. Secondly, the
NMPA is trying to further simplify the application procedure for class II
devices; in addition to reducing the unnecessary cost, time and effort the
applicants have to invest during the preparation phase, this could also lead to
shorter review time for class II devices. Such positive attitude from the NMPA
is definitely good news to all applicants, and especially to manufacturers of
class II medical devices.
4.3 Alternative options to registration testing
The 2017 proposal of "Regulations for the Supervision and Administration of
Medical Devices (draft version)" also includes the revisions to the
documentation requirements on the submission dossier, in particular on the
requirement of product test report (investigation of product’s characteristics
versus corresponding standards) and clinical evaluation. Table 13 shows the
differences between the proposed version and the current version (note:
translated into English by the author of this master thesis).
"Regulations for the Supervision and Administration of Medical Devices"
Current version (2014)
Proposed version (2017)
Article 9:
The following documentation should be
submitted for the filing of class I medical
devices and the registration application
of class II and class III medical devices:
1) risk analysis documentation;
2) product technical requirement;
3) product test report;
4) clinical evaluation
documentation;
5) product IFU and label sample;
6) quality system documentation
Article 9:
The following documentation should be
submitted for the filing of class I medical
devices and the registration application
of class II and class III medical devices:
1) risk analysis documentation;
2) product technical requirement;
3) product test report;
4) product IFU and label sample;
5) quality system documentation
related to product development and
manufacture;

4. Discussion
37
related to product development and
manufacture;
7) other data to demonstrate the
product safety and efficacy.
The applicant is responsible for the
truthfulness of the submitted
documentation.
6) other data to demonstrate the
product safety and efficacy.
The product test report may be the
contract testing report from a qualified
test institution or self-testing report
from the applicant or filing entity.
For applications of class II and class III
medical devices that require clinical
evaluation, the clinical evaluation report
should be submitted.
The applicant is responsible for the
truthfulness, completeness and
traceability of the submitted
documentation.
Article 10
For class I medical devices, the product
test report can be self-testing report from
the applicant or filing entity; the clinical
evaluation documentation does not
require clinical trial report…
…
Article 10
(integrated into Article 9)
…
Article 11
…
For class II and class III medical devices,
the product test report should be
registration testing report from a qualified
test institution; the clinical evaluation
documentation should include clinical
trial report unless exempted.
Article 11
…
(integrated into Article 9)
Table 13: Comparison of requirements on submission dossier
The revision on the requirements of clinical evaluation is in line with the
proposed exemption of clinical trial for class II devices and further simplifies
the documentation.
The seemingly simplification on the requirements of the registration test does
not necessarily mean the exemption of registration testing. Table 14 shows
the requirements on registration testing from both the replaced version and
current version of the "Provisions for Medical Device Registration" (note:
translated into English by the author of this master thesis).

4. Discussion
38
"Provisions for Medical Device Registration"
Replaced version (2004)
Current version (2014)
Article 9
Class II and class III medical devices
have to complete registration testing
conducted by the test institutions
qualified by both the State Food and
Drug Administration and the General
Administration of Quality Supervision,
Inspection and Quarantine of the
People's Republic of China.
Clinical trials or registration application
can be carried out only after the
registration testing confirmed that the
product complies with the product
standard.
A list of qualified institutions will be
published separately.
Article 16
When applying for class II and class III
medical device registration, registration
testing shall be conducted. The medical
device testing institutions shall carry out
relevant registration testing on the
products in accordance with the product
technical requirements.
The production of samples for
registration testing shall comply with
relevant requirements of quality
management system for medical
devices. Clinical trials or registration
application can be carried out only after
the products passed the registration
testing.
For the filing of class I medical device,
the filing entity may submit the product
self-testing report.
Article 18
Medical device testing institutions shall
have the relevant qualification of medical
device testing, and perform testing within
their specified testing scope. It shall
carry out pre-evaluation of the product
technical requirements. The pre-
evaluation opinions together with the
medical device registration testing report
shall be issued to the applicant.
For the medical devices having not
included in the testing scope of any
medical device testing institutions, the
corresponding registration department
shall designate a testing institution which
has the capability to conduct the testing.
Table 14: Requirements on registration testing
Considering that the registration test report has always been the crucial and
mandatory document for medical device applications in China, and the test
institutions have to be qualified by the authorities, it is in the opinion of the

4. Discussion
39
author of this master thesis very unlikely that self-testing reports can be used
to replace the registration testing report for class II and class III medical
device applications. Additionally, the test institution that conducts the
registration testing plays the very important role as the pre-reviewers of the
PTR (replaced the previous product standard) as designated by the
regulations. The pre-evaluation should cover the following aspects (note:
translated into English by the author of this thesis):
[30]
the completeness and applicability of the performance criteria; the
feasibility, repeatability and the suitability of the testing method;
the completeness and applicability of the mandatory national standard
and industrial standard and the suitability of the clauses being referred
to;
the feasibility, repeatability and the suitability of the referenced content
from the China Pharmacopeia.
As the pre-evaluation should cover many China specific requirements, in
case of self-testing reports, especially done by foreign manufacturers, there
could be issues with the acceptability of the PTR.
Since April 2017 the registration testing fee has been cancelled
[31]
, leading to
long waiting time for registration testing procedures and delay of applications.
This issue has been noticed by the CFDA already, stating that contract
testing report will also be accepted for registration purpose, as long as the
test has been conducted in accordance with the PTR and a pre-evaluation
conclusion from the test lab has been included.
[32]
Considering that both the
registration test and contract test are type testing conducted by qualified test
institutions, and the pre-evaluation conclusion from the test lab is still a
mandatory document for submissions in China, the proposed revision on the
registration test requirement could be the attempt from the NMPA to accept
other form of type testing for registration applications, rather than the sign of
exempting type testing in China.
4.4 Post market surveillance being strengthened
According to Article 5 of the “Provisions for Adverse Events Surveillance and
Re-assessment of Medical Devices”, the NMPA will establish a national
surveillance system for adverse events of medical devices. Article 14 of the
same “Provisions” requires the applicant to establish a quality management

4. Discussion
40
system that includes adverse events surveillance and product re-assessment;
the adverse events should be collected and reported in time and the product
risk evaluation report should be updated periodically based on surveillance
data. The trend of strengthening on medical device surveillance in China is in
line with the trend in the EU. However, it is worth noting that adverse events
occurred both within and outside China are required to be reported to the
NMPA for imported devices. Therefore, if the products are being marketed
both in China and in the EU, it would make sense to cover the new specific
requirements in China when updating the post market surveillance system to
meet the new MDR requirement.
Requirements for the “Marketing Approval Holders” (MAHs) of imported
medical devices to report adverse events occurred outside China:
Serious adverse events leading to the product being controlled should
be reported to the NMPA within 24 h after the applicant being informed;
Serious adverse events that may lead to serious injury or death of the
patient should be reported to the NMPA within 30 days after the
applicant being informed.
Additionally, the MAHs of imported medical devices should submit a
periodical risk evaluation report to the NMPA according to the regulations,
which should summarize and analyse the adverse events and surveillance
data from both within and outside China to re-evaluate the risk and benefit of
the product and record the risk control measures being taken.
According to the “Provisions for Recalls of Medical Devices”
[33]
,
the MAHs of
imported medical devices should report recalls conducted outside China to
the NMPA as well.
4.5 Overseas on-site audits to be conducted
Since December 2015 the CFDA started the implementation of overseas on-
site audits. Reports from the 24 overseas on-site audits conducted in 2017
have been published by the CFDA on its website. According to the NMPA, 26
overseas on-site audits have been conducted in 2018 and all the audit
reports will be published on its website. So far, 5 reports from the overseas
on-site audits conducted in 2018 have been published already.
[34]

4. Discussion
41
In China, the harmonized standard ISO 13485:2016
(3)
has been adapted into
a recommended industrial standard YY / T 0287-2017
( 4 )
and is not
mandatory. The mandatory QMS regulation in China and the basis for
overseas on-site audit is the “Good Manufacture Practice for Medical Devices”
[35]
(including its annexes
[36]
[37]
[38]
when applicable), covering the design,
manufacture, sales and after sale services of medical devices to be and
being marketed in China. In general, the “Good Manufacture Practice for
Medical Devices” has no conflict with the GMP or ISO 13485. Because the
on-site audit is to be conducted selectively, for initial application for
registration of imported medical devices, the NMPA will accept the QMS
qualification of the foreign applicants issued by the country of origin.
Concerning applicants from the EU (including UK), an ISO 13485 certificate
will be accepted as the manufacturer’s qualification by the NMPA. However,
special attention should be paid to the China specific requirements that
should be covered by the QMS for medical devices being or to be marketed
in China. For example, the PTR and applicable Chinese standards should be
included in the technical documentation according to Article 24 of the “Good
Manufacture Practice for Medical Devices”. And to study the overseas on-site
audit reports published by the CFDA is highly recommended. Among these
29 reports, the author of this master thesis found that aside from common
QMS non-conformities, other common non-conformities specific to Chinese
regulations have been found, including the items listed in Table 15 below.
Non-conformity items
Reports in
total
Chinese regulations not being (correctly) identified
[40] [41] [42] [43] [44]
[45] [46] [47] [49] [50] [52] [53]
12
a
Inconsistencies with the PTR / product standard
[39] [41] [43] [46] [51] [52]
[54]
7
b
Specifications / components being marketed in China without
CFDA approval
[39] [49]
2
c
(3)
ISO 13485:2016, Medical devices -- Quality management systems -- Requirements for
regulatory purposes
(4)
YY/T 0287-2017, Medical devices — Quality management systems — Requirements for
regulatory purposes (ISO 13845:2016, IDT*)
*IDT = identical

4. Discussion
42
Chinese IFU and label not meeting CFDA requirements
[42]
1
Clean room condition inconsistent with Chinese GMP
[48]
1
Notes:
a) 7 reports on the manufacturers based in the EU (including the UK);
b) 6 reports on the manufacturers based in the EU (including the UK);
c) Both reports on the manufacturers based in the UK.
Table 15: Non-conformities to Chinese regulations
Considering that the ISO 13485 certificate from the manufacturer based in
the EU (including the UK) is the mandatory qualification file to be submitted
for applications of imported medical devices, the non-conformities listed in
Table 15 are good examples that the compliance with ISO 13485 will not be
enough for the Chinese “Good Manufacture Practice for Medical Devices”
and other related Chinese regulations. The European manufacturers have to
identify applicable Chinese regulations as early as possible to ensure the
regulatory compliance of their medical devices to be and being marketed in
China.
It is worth noting that Chinese local medical device manufacturers have to
pass the on-site QMS audit before submitting the applications for their
products, while most of the medical devices manufacturers overseas have
never been audited on site by the CFDA. The non-execution of on-site QMS
audits to overseas medical device manufacturers is largely due to the
restrictions from practical and political aspects, such as manpower and visa
application. So far, China has no plan to join the “Mutual Recognition
Agreements” or accept audit reports issued by conformity assessment bodies
from other countries. Even though in reality the overseas on-site audits have
to be done selectively, the medical device manufacturers have the
responsibility to comply with Chinese regulations when marketing their
products in China. Hence, the urgency of identifying and adopting Chinese
regulations should not be measured by the “risk” of being selected for the
overseas on-site audit.
4.6 Special review procedure for innovative devices
For initial registration application of imported medical devices in China, the
approval from the country of origin has always been the basic requirement; it

4. Discussion
43
is the mandatory document to be submitted as requested by both the
previous (2004) version and current (2014) version of the “Provisions for
Medical Device Registration”, and by the current (2014) version of the
“Regulations for the Supervision and Administration of Medical Devices”. A
positive sign from the proposed “Regulations for the Supervision and
Administration of Medical Devices (draft version)” is the exemption on the
marketing approvals from the country of origin (= valid certificate of a Notified
Body for European applicants) for innovative medical devices. However,
there are many restrictions on the qualification as an “innovative” device.
Table 16 below shows the provisions concerning the requirements of
marketing approval from the country of origin in the current version (2014)
and proposed draft (2017) of the “Regulations for the Supervision and
Administration of Medical Devices” (note: translated into English by the
author of this master thesis).
"Regulations for the Supervision and Administration of Medical Devices"
Current version (2014)
Proposed version (2017)
Article 11:
…
For applications of imported class II and
class III medical devices, the marketing
approval from the country of origin for
the product to be marketed in China
should be submitted to the CFDA by the
agent.
…
Article 11:
…
For applications of imported class II and
class III medical devices, the marketing
approval from the country of origin for
the product to be marketed in China
should be submitted to the NMPA by the
agent. For innovative medical devices
that have not been marketed outside
China yet, the marketing approval from
the country of origin can be exempted.
…
Table 16: Requirement on marketing approval from the country of origin
As the proposal showed already, the exemption will only apply to devices that
have not been put on the market yet. Additionally, the NMPA also tightened
the eligibility criteria to narrow down the applicants. In 2018 the NMPA issued
the final version on the special review procedure of innovative medical
devices, further restricting the qualified innovative medical devices to medical
devices that have a Chinese patent and the qualification application should

4. Discussion
44
be submitted within 5 years after the publish date of its Chinese patent. The
qualification of the patent will be reviewed during the expert meetings.
Despite all the restrictions, the proposed exemption on the marketing
approval from the country of origin is still a positive sign that China is trying to
show more favour to innovative medical devices. And this is not the first
positive sign. In 2014 the CFDA has already issued a draft on the special
review procedure of innovative medical devices, enabling earlier interaction
with the reviewer and reducing queuing time during the testing and review
procedures for qualified innovative medical devices. Although such special
procedure will be good news to manufacturers of innovative medical devices
who want to market their products in China as soon as possible, the
procedure itself emphasizes that the technical requirements will not be
compromised and the necessary procedures will not be omitted, meaning
that if the foreign manufacturers wants to put their innovative medical device
onto the market of China soon, they should be prepared for the requests on
conducting thorough tests in China; and while the queuing time during the
testing and review procedures will be shortened, the actual procedures of the
testing and reviews could not be accelerated. In fact, whenever expert
meetings are involved, the technical review would most likely take longer
time and additional testing requests are to be expected. Even so, the chance
of early interaction with the reviewer and experts will be really helpful to
optimise the test design and avoid unnecessary cost and time during the
preparation and review phases.

5. Conclusion and outlook
45
5. Conclusion and outlook
Although compared to medical device regulations in the EU, the Chinese
medical device regulations are relatively immature and are being revised
more frequently, the trend indicates that China is attempting to simplify the
application procedures and be more open to new medical technologies while
strengthening on the post market surveillance.
For many years, in China, the marketing approval of a medical device would
be largely based on local testing data, including local type testing data and
local clinical trial data. Although it will take time to review the system to
accept respectively to implement alternative approaches, the up to now
respectively amended and new elaborated regulations have already shown
positive signs. Now being a regular member of the ICH as well as a full
member of the IMDRF, the NMPA actively tries to accelerate the procedure
to adopt harmonized standards and get in line with international regulations.
It is to be expected that the discrepancies between Chinese and EN ISO (=
European and International) standards and EU and FDA regulations will be
less significant in the future.
Regarding post market surveillance, now that the MDR also strengthens the
requirements for medical devices being marketed in the EU, to improve the
plan, implementation and documentation of the post market surveillance
activities will be the major tasks of European manufacturers. Hence the gap
between the requirements on post market surveillance in the EU and in
China should be less significant if the manufacturer complies with the MDR.
Although the signs are positive, the reform of the medical device regulation
system in China will definitely take time. And considering the economic and
political differences, China specific requirements will still exist. For European
medical device manufacturers that prepare to enter the market of China or
have already been marketing their products in China, it is recommendable to
keep up with the development of Chinese medical device regulations. The
early and correct input of Chinese regulations will be the best approach to
avoid unnecessary time, cost and effort in later stages. Identifying the correct
Chinese requirement in the design phase will be more cost-effective than

5. Conclusion and outlook
46
trying to cover the gap later during the application phase (for details refer to
chapter 4 of this master thesis).
Because the medical device regulations, guidelines and standards in China
will be issued in Chinese only, language could be an issue to European
manufacturers. However, it is not impossible for any foreign manufacturer to
keep up with the new Chinese regulations, as long as the responsibilities of
the agent have been clearly defined and the communication between the
agent and the foreign manufacturer is efficient enough.
Table 17 and Table 18 below list the latest version of some very important
Chinese regulations and helpful guidelines for medical devices.
Regulations
Issue date
Effective
date
Regulations for the Supervision and Administration of
Medical Devices (Decree No. 650 of the state council)
2014-03-07
2014-06-01
Provisions for Medical Device Registration (Decree No.
4 of the CFDA)*
2014-07-30
2014-10-01
Provisions for Instructions and Labels of Medical
Devices (Decree No. 6 of the CFDA)*
2014-07-30
2014-10-01
Good Manufacture Practice for Medical Devices
2014-12-29
2015-03-01
Good Clinical Practice for Medical Devices (Decree
No. 25 of the CFDA)
2016-03-01
2016-06-01
Provisions for Recalls of Medical Devices (Decree
No.29 of the CFDA)
2017-01-25
2017-05-01
Rules for Classification of Medical Devices (Decree
No.15 of the CFDA)*
2015-07-14
2016-01-01
Medical Device Classification Catalogue
2017-08-31
2018-08-01
Provisions for Adverse Events Surveillance and Re-
assessment of Medical Devices” (Decree No. 1 of the
SAMR)
2018-08-13
2019-01-01
* English version available. Links given in “References”.
Table 17: List of important Chinese regulations
Guidelines
Issue date
Guideline on drafting Product Technical Requirement
2014-05-10
Guideline on the Clinical Evaluation of Medical Devices
2015-04-01
Guideline on Division of Registration Unit
2017-11-17

5. Conclusion and outlook
47
Guideline on Accepting Foreign Clinical Data
2018-01-10
Guideline on Initial Registration of Imported Medical Devices
2018-09-30
Guideline on Change Application of Imported Medical Devices
2018-09-30
Guideline on Renewal Application of Imported Medical Devices
2018-09-30
Table 18: List of useful Chinese guidelines

6. Summary
48
6. Summary
While being an attractive market to foreign medical device manufacturers,
China is also considered to be one of the difficult markets due to the Chinese
specific and rapidly changing regulations. Especially since the former State
Food and Drug Administration (SFDA) reconstructed into the China Food and
Drug Administration (CFDA) in 2013, the medical device regulations in China
went through many changes, including major ones. In 2014, two fundamental
regulations, the "Regulations for the Supervision and Administration of
Medical Devices"
and the "Provisions for Medical Device Registration" have
been revised and came into force. Subsequently other regulations have been
revised and new regulations and guidelines have been issued as well,
providing clarifications to critical issues such as the requirement on clinical
evaluations. The CFDA attempted to better normalize the review and
approval procedures for medical device applications.
In March 2018 the CFDA has been reconstructed into the National Medical
Products Administration (NMPA), which is now under the supervision of the
State Administration for Market Regulation (SAMR). The post market
surveillance of medical devices in China will be strengthened. New changes
to the medical devices regulations in China are to be expected.
This master thesis took a general but systematic review of the current
medical device regulations in China that have come into force since 2014,
analysing the gap between the regulatory requirements in China and in the
EU, with the focus on application procedures. This thesis also identified some
of the most intriguing new trend in the recent changes of Chinese medical
device regulations and discussed the possible challenges and opportunities
to be faced by European medical device manufacturers that comply already
with the medical device regulations in the EU. According to the author’s
opinion, even though China will remain challenging to foreign manufacturers
for quite some time, the recent changes of Chinese medical device
regulations indicated positive signs of better normalized procedures.
Although language could be an issue, as long as the foreign manufacturers
manage the communication with their local agents effectively, it’s not
impossible to keep up with the regulation changes in China. Besides,
identifying the local specific requirements as early as possible will definitely

6. Summary
49
be more helpful than trying to cover the gap later, which applies to any other
markets as well.

Acknowledgement
VI
Acknowledgement
I'm extremely grateful to my supervisor Dr. Ehrhard Anhalt for his time and
patience, and for his insightful comments during the review of this master thesis.
I'm also grateful to Dr. Santiago Figueroa-Perez for taking the time to review this
master thesis.
Additionally, I would like to thank Ms. Barbara Röcher, Ms. Jasmin Fahnenstich,
Ms. Eva-Maria Eibl and Ms. Charlotte Klement for their helpful support and
excellent organization during the courses.

References
VII
References
[1] State Council of the People's Republic of China. (2014). Regulations for the Supervision
and Administration of Medical Devices (Decree No. 650 of the state council),
http://samr.cfda.gov.cn/WS01/CL1100/97814.html. Accessed on 3 January 2019. (only
Chinese version available)
[2] CFDA. (2014). Provisions for Medical Device Registration (Decree No. 4 of the CFDA),
http://eng.sfda.gov.cn/WS03/CL0768/144282.html. Accessed on 3 January 2019.
[3] IMDRF. (2013). Outcome Statement of the IMDRF-3 Management Committee, 19 to 21
March 2013, http://www.imdrf.org/docs/imdrf/final/meetings/imdrf-meet-130319-france-
outcome-statement.pdf. Accessed on 3 January 2019.
[4] ICH. (2017). Minutes - ICH Management Committee Meeting - Montreal, Canada - May
29 - June 1, 2017,
https://www.ich.org/fileadmin/Public_Web_Site/Meetings/Ass_MC_Meetings_Reports/M
inutes_MC_ICH_Meeting_Montreal_May2017.pdf. Accessed on 3 January 2019.
[5] CFDA. (2014). Announcement on publishing the “Special Review Procedure of
Innovative Medical Devices (interim version)” (No. 13 Announcement of 2014),
http://www.nmpa.gov.cn/WS04/CL2197/324797.html. Accessed on 7 January 2019.
(only Chinese version available)
[6] CFDA. (2017). Seeking public comment on the “Guideline on Accepting Foreign Clinical
Data (draft version)”, http://www.cmde.org.cn/CL0066/6826.html. Accessed on 7
January 2019. (only Chinese version available)
[7] CFDA. (2018). CFDA announcement of publishing the “Guideline on Accepting Foreign
Clinical Data” (No. 13 Announcement of 2018),
http://samr.cfda.gov.cn/WS01/CL2039/222385.html. Accessed on 3 January 2019. (only
Chinese version available)
[8] CFDA. (2017). Regulations for the Supervision and Administration of Medical Devices
(draft version), http://www.moj.gov.cn/government_public/content/2018-
06/25/tzwj_21120.html. Accessed on 7 January 2019. (only Chinese version available)
[9] SAMR. (2018). “Provisions for Adverse Events Surveillance and Re-assessment of
Medical Devices” (Decree No. 1 of the SAMR),
http://www.nmpa.gov.cn/WS04/CL2077/330071.html. Accessed on 3 January 2019.
(only Chinese version available)
[10] NMPA. (2018). Announcement on issuing the “Special Review Procedure of Innovative
Medical Devices” (No. 83 Announcement of 2018),
http://www.nmpa.gov.cn/WS04/CL2138/331560.html. Accessed on 7 January 2019.
(only Chinese version available)
[11] NMPA. (2018). Seeking public comment on the “Provisions for the Supervision and
Administration of Agents of Imported Medical Devices (draft version)”,
http://www.nmpa.gov.cn/WS04/CL2051/329805.html. Accessed on 3 January 2019.
(only Chinese version available)

[12] CFDA. (2015). Rules for Classification of Medical Devices (Decree No.15 of the CFDA),
http://eng.sfda.gov.cn/WS03/CL0768/144302.html. Accessed on 3 January 2019.
[13] CFDA. (2017). CFDA announcement of publishing the “Medical Device Classification
Catalogue” (No. 104 Announcement of 2017),
http://samr.cfda.gov.cn/WS01/CL1294/177089.html. Accessed on 3 January 2019. (only
Chinese version available)
[14] Yuan Peng. (2017). Update on Medical Device regulatory in China,
http://www.imdrf.org/docs/imdrf/final/meetings/imdrf-meet-170919-canada-presentation-
jurisdictional-update-china.pdf. Accessed on 3 January 2019.
[15] CFDA. (2017). CFDA announcement of publishing the “Guideline on Division of
Registration Unit” (No. 187 Announcement of 2017).
http://samr.cfda.gov.cn/WS01/CL0087/217314.html. Accessed on 3 January 2019. (only
Chinese version available)
[16] CFDA. (2014). CFDA announcement of the requirement on submission documents and
the format of certificates (No. 43 Announcement of 2014),
http://samr.cfda.gov.cn/WS01/CL0087/106095.html. Accessed on 3 January 2019. (only
Chinese version available)
[17] CFDA. (2014). Provisions for Instructions and Labels of Medical Devices (Decree No. 6
of the CFDA), http://eng.sfda.gov.cn/WS03/CL0768/144301.html. Accessed on 3
January 2019.
[18] CFDA. (2015). CFDA announcement of publishing the procedure of reissuing medical
device registration certificates and other procedures (No. 91 Announcement of 2015),
http://www.nmpa.gov.cn/WS04/CL2183/322088.html. Accessed on 3 January 2019.
(only Chinese version available)
[19] CMDE. (2018). Flowchart of application procedures, https://www.cmde.org.cn/CL0118/.
Accessed on 3 January 2019. (only Chinese version available)
[20] NMPA. (2018). Guideline on Initial Registration of Imported Medical Devices,
http://samr.cfda.gov.cn/WS01/CL1768/152057.html. Accessed on 3 January 2019. (only
Chinese version available)
[21] NMPA. (2018). Guideline on Change Application of Imported Medical Devices,
http://samr.cfda.gov.cn/WS01/CL1768/152056.html. Accessed on 3 January 2019. (only
Chinese version available)
[22] NMPA. (2018). Guideline on Renewal Application of Imported Medical Devices,
http://samr.cfda.gov.cn/WS01/CL1768/152055.html. Accessed on 3 January 2019. (only
Chinese version available)
[23] GHTF. (2008). Summary Technical Documentation for Demonstrating Conformity to the
Essential Principles of Safety and Performance of Medical Devices (STED),
http://www.imdrf.org/docs/ghtf/final/sg1/technical-docs/ghtf-sg1-n011-2008-principles-
safety-performance-medical-devices-080221.pdf. Accessed on 3 January 2019.

[24] CFDA. (2014). Guideline on drafting Product Technical Requirement (No. 9
Announcement of 2014), https://www.cmde.org.cn/CL0112/7877.html. Accessed on 3
January 2019. (only Chinese version available)
[25] CMDE. (2015). Guideline on the Clinical Evaluation of Medical Devices,
http://www.cmde.org.cn/CL0112/6152.html. Accessed on 3 January 2019. (only
Chinese version available)
[26] CFDA. (2018). CFDA announcement on the implementation of pre-submission
consultation (No. 4 Announcement 2018), https://www.cmde.org.cn/CL0004/7970.html.
Accessed on 3 January 2019. (only Chinese version available)
[27] CFDA. (2018). CFDA announcement on the revision of documentation requirement on
the renewal application, etc. (No. 53 Announcement of 2018),
http://www.nmpa.gov.cn/WS04/CL2050/329976.html. Accessed on 3 January 2019.
(only Chinese version available)
[28] CFDA. (2016). Good Clinical Practice for Medical Devices (Decree No. 25 of the CFDA),
http://www.nmpa.gov.cn/WS04/CL2186/300685.html. Accessed on 3 January 2019.
(only Chinese version available)
[29] ICH. (2016). Integrated Addendum to ICH E6 (R1): Guideline for Good Clinical Practice
- E6 (R2),
https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6
_R2__Step_4_2016_1109.pdf. Accessed on 3 January 2019.
[30] CFDA. (2014). CFDA announcement on the request of pre-evaluation of the product
technical requirement to be conducted by medical device test institutions (No. 192
Announcement of 2014), http://www.nmpa.gov.cn/WS04/CL2197/324832.html.
Accessed on 6 January 2019. (only Chinese version available)
[31] Ministry of Finance of the People's Republic of China. (2017). Announcement on the
cancellation of certain administration fees,
http://www.mof.gov.cn/gp/xxgkml/szs/201703/t20170323_2563593.html. Accessed on 6
January 2019. (only Chinese version available)
[32] CFDA. (2017). CFDA announcement on the implementation of medical device testing
and related works (No. 187 Announcement of 2017),
http://www.nmpa.gov.cn/WS04/CL2187/322919.html. Accessed on 6 January 2019.
(only Chinese version available)
[33] CFDA. (2017). Provisions for Recalls of Medical Devices (Decree No.29 of the CFDA),
http://www.nmpa.gov.cn/WS04/CL2186/300689.html. Accessed on 6 January 2019.
(only Chinese version available)
[34] NMPA. (2018). NMPA published reports from overseas on-site audits of 5 imported
medical device manufacturers, http://www.nmpa.gov.cn/WS04/CL2176/334555.html.
Accessed on 18 January 2019. (only Chinese version available)

[35] CFDA. (2014). CFDA announcement on publishing the “Good Manufacture Practice for
Medical Devices” (No. 64 Announcement of 2014),
http://www.nmpa.gov.cn/WS04/CL2138/299995.html. Accessed on 7 January 2019.
(only Chinese version available)
[36] CFDA. (2015). CFDA announcement on publishing the Annex “Sterile Medical Devices”
of the “Good Manufacture Practice for Medical Devices” (No. 101 Announcement of
2015), http://www.nmpa.gov.cn/WS04/CL2183/322078.html. Accessed on 7 January
2019. (only Chinese version available)
[37] CFDA. (2015). CFDA announcement on publishing the Annex “Implantable Medical
Devices” of the “Good Manufacture Practice for Medical Devices” (No. 102
Announcement of 2015), http://www.nmpa.gov.cn/WS04/CL2183/322079.html.
Accessed on 7 January 2019. (only Chinese version available)
[38] CFDA. (2015). CFDA announcement on publishing the Annex “In vitro Diagnostics” of
the “Good Manufacture Practice for Medical Devices” (No. 103 Announcement of 2015),
http://www.nmpa.gov.cn/WS04/CL2183/322080.html. Accessed on 7 January 2019.
(only Chinese version available)
[39] CFDA. (2017). Notification on the overseas on-site audit report - Leica Biosystems
Newcastle Ltd, http://samr.cfda.gov.cn/WS01/CL2026/219208.html. Accessed on 7
January 2019. (only Chinese version available)
[40] CFDA. (2017). Notification on the overseas on-site audit report - MedTrade Products
Limited, http://samr.cfda.gov.cn/WS01/CL2026/217547.html. Accessed on 7 January
2019. (only Chinese version available)
[41] CFDA. (2018). Notification on the overseas on-site audit report - Metrax GmbH,
http://samr.cfda.gov.cn/WS01/CL2026/222858.html. Accessed on 7 January 2019. (only
Chinese version available)
[42] CFDA. (2018). Notification on the overseas on-site audit report - NovaBone Products,
LLC, http://samr.cfda.gov.cn/WS01/CL2026/225516.html. Accessed on 7 January 2019.
(only Chinese version available)
[43] CFDA. (2017). Notification on the overseas on-site audit report - PRODIMED,
http://samr.cfda.gov.cn/WS01/CL2026/217548.html. Accessed on 7 January 2019. (only
Chinese version available)
[44] CFDA. (2018). Notification on the overseas on-site audit report - Sewoon Medical Co.,
Ltd, http://samr.cfda.gov.cn/WS01/CL2026/222856.html. Accessed on 7 January 2019.
(only Chinese version available)
[45] CFDA. (2018). Notification on the overseas on-site audit report - STAR Medical Co., Ltd,
http://samr.cfda.gov.cn/WS01/CL2026/222857.html. Accessed on 7 January 2019. (only
Chinese version available)
[46] CFDA. (2018). Notification on the overseas on-site audit report - William A. Cook
Australia Pty, Ltd., http://samr.cfda.gov.cn/WS01/CL2026/225518.html. Accessed on 7
January 2019. (only Chinese version available)

[47] CFDA. (2018). Notification on the overseas on-site audit report - 3M Health Care,
http://samr.cfda.gov.cn/WS01/CL2026/225517.html. Accessed on 7 January 2019. (only
Chinese version available)
[48] CFDA. (2018). Notification on the overseas on-site audit report - Angiomed GmbH
Co.Medizintechnik KG, http://samr.cfda.gov.cn/WS01/CL2026/222855.html. Accessed
on 7 January 2019. (only Chinese version available)
[49] CFDA. (2017). Notification on the overseas on-site audit report - Biocomposites Ltd,
http://samr.cfda.gov.cn/WS01/CL2026/219209.html. Accessed on 7 January 2019. (only
Chinese version available)
[50] CFDA. (2017). Notification on the overseas on-site audit report - BIOKIT, S.A.,
http://samr.cfda.gov.cn/WS01/CL2026/217544.html. Accessed on 7 January 2019. (only
Chinese version available)
[51] CFDA. (2017). Notification on the overseas on-site audit report - LABORATOIRES
URGO, http://samr.cfda.gov.cn/WS01/CL2026/217544.html. Accessed on 7 January
2019. (only Chinese version available)
[52] NMPA. (2018). Notification on the overseas on-site audit report - Rayner Intraocular
Lenses Limited, http://www.nmpa.gov.cn/WS04/CL2195/334552.html. Accessed on 18
January 2019. (only Chinese version available)
[53] NMPA. (2018). Notification on the overseas on-site audit report - OPHTEC B.V.,
http://www.nmpa.gov.cn/WS04/CL2195/334552.html. Accessed on 18 January 2019.
(only Chinese version available)
[54] NMPA. (2018). Notification on the overseas on-site audit report - Becton Dickinson and
Company, http://www.nmpa.gov.cn/WS04/CL2195/334534.html. Accessed on 18
January 2019. (only Chinese version available)
Erklärung
Hiermit erkläre ich an Eides statt, die Arbeit selbständig verfasst und keine
anderen als die angegebenen Hilfsmittel verwendet zu haben.
__________________________
